
Written by:
Núria Nieto, Specialist in Advanced Therapies at Klinea Biotech & Pharma Engineering
Jordi Gibert, Head of the Biotechnology Unit at Klinea Biotech & Pharma Engineering
Ander Izeta, Head of Section – Advanced Therapies Unit, Donostia University Hospital
Advanced therapy medicinal products (ATMPs) are transforming modern medicine. From CAR-T therapies[1], which have turned certain refractory lymphomas into treatable diseases, to bioengineered corneal implants, which have restored vision in blind patients[2], these types of treatments are no longer experimental but have become real clinical options. Autologous CAR-T therapies are produced and administered in European public hospitals under hospital exemption schemes, expanding their reach without relying solely on industry[4].
However, this revolution poses complex challenges across the entire value chain (Figure 1): from production under Good Manufacturing Practices (GMP), logistics and storage, to regulatory and ethical access. The lack of regulatory harmonisation, limited specialised training and high costs hinder its scalability. At the same time, emerging technologies such as dynamic cultivation systems, artificial intelligence and biobanks open up new opportunities (Figure 2), but also generate regulatory uncertainties[4].
Technical and manufacturing challenges
One of the main obstacles lies in the transition from preclinical research environments to industrial production systems (GMP). This change is neither linear nor straightforward, and involves technical, logistical, and regulatory compliance obstacles that directly affect scalability and equity of access[5].
The ‘living’, heterogeneous and personalised nature of ATMPs imposes unique barriers compared to conventional medicines. In the case of autologous therapies, each batch is manufactured from the biological material of a single patient, which transforms production into an almost artisanal process. Cellular variability between individuals prevents effective standardisation, complicating quality control and consistency between batches[4]. Although the implementation of closed platforms has enabled advances towards automation[6], their adoption requires substantial investment and highly trained personnel, limiting their effective integration into public hospitals.
In the production phase, the challenges begin with obtaining suitable biological material. Isolating viable cells from the patient themselves is a demanding and time-consuming process, often affected by multiple previous treatments. Furthermore, in gene therapies, the production of viral vectors is costly and relatively inefficient, affecting product availability and representing another impediment to the application of the therapy. Added to this are logistical complexities, such as the transfer of cells between collection and production centres, which cause significant delays in batch manufacturing and may delay access to commercial therapy for patients who have been approved for treatment[4].
During distribution, products must be transported under extreme cryogenic conditions, which involves the use of deep-freezing technologies and cryoprotective agents such as DMSO, which can affect cell viability and are toxic if not handled correctly[4]. In addition, clinical administration requires advanced infrastructure for sterile handling, vein-to-vein traceability[4] and rapid response to potential adverse effects. All of this limits the expansion of these therapies to jurisdictions whose health systems have less installed capacity.
Finally, quality controls must guarantee the sterility, cell viability and stability of the product, as well as the absence of microbial contaminants or endotoxins, requiring advanced analytical protocols and rigorous validation processes. In the case of pluripotent cell-based therapies, specific risks such as tumorigenicity must also be mitigated, which requires highly sensitive functional assays[4]. This calls for advanced assays, many of which require cross-validation between GLP and GMP environments, representing a critical point of technical and regulatory vulnerability.
Regulatory challenges
Despite regulatory advances, such as Regulation 1394/2007[7] in Europe, there remains significant regulatory fragmentation between different countries. The lack of international harmonisation slows down the global development of therapies and complicates their multiregional approval. Cases such as Holoclar®, authorised by the European Medicines Agency (EMA) in 2015[8] but not yet approved by the Food and Drug Administration (FDA)[9], exemplify these asymmetries.
One critical point is decentralised manufacturing. The hospital exemption, regulated by European Regulation No. (EC) 1394/2007[7] and Royal Decree 477/2014[10] in Spain, allows hospitals to produce ATMPs without going through the centralised procedure, under the supervision of their national authority. Although this route has proven effective for personalised and low-volume therapies, its implementation varies between EU countries[11], introducing legal uncertainty, unequal opportunities for public developers and difficulties in access for patients in different regions[12].
On the other hand, the high cost of these therapies poses a structural problem. The price of commercial CAR-T therapy ranges between €300,000 and €400,000 per patient, placing strain on the financial sustainability and equity of public health systems [13]. The response has been partial: pay-for-performance models have been adopted in countries such as Italy and Spain, but their adoption is limited and is also accompanied by opacity in bureaucratic agreements [14]. Meanwhile, public-academic initiatives such as ARI-0001 at the Hospital Clínic de Barcelona, with costs of
€90,000 [15]. or NC-1 at the Hospital Puerta de Hierro in Majadahonda, show that it is possible to offer viable alternatives with lower costs and greater public control [16].
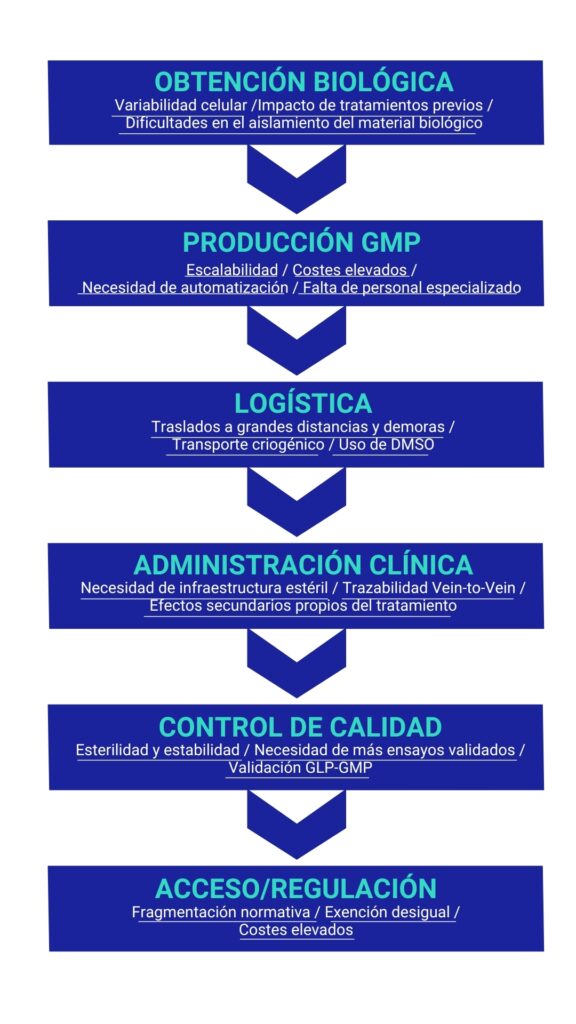
Figure 1. Value chain and challenges of ATMPs
Evidence, predictability, and security
The responsible deployment of ATMPs requires strengthening evidence generation and post-authorisation surveillance. However, there is still no comprehensive pan-European system that systematically collects longitudinal data on safety, efficacy and health outcomes for all ATMPs. This absence limits the ability of regulators to make decisions based on real data. A notable exception is the European Society for Blood and Marrow Transplantation (EBMT) registry, which currently includes nearly 15,000 CAR-T cell treatments and is playing an essential role in generating long-term clinical evidence in this field[17].
Furthermore, the lack of predictability of current preclinical models, both in vitro and animal, leads to high failure rates in clinical phases, making development more expensive. Investment in patient-derived organoids, organ-on-chip models, and artificial intelligence-based in silico simulations is essential to improve clinical translation and reduce late failures in clinical phases[4]. One example of success in this regard is the use of organoids to predict the efficacy of personalised treatment in cystic fibrosis[18].
Another growing challenge is the proliferation of unregulated therapies offered by private clinics in environments with little oversight. These pseudo-therapies, lacking scientific validation, pose a serious risk to patients and undermine confidence in advanced therapies. Addressing this phenomenon requires international coordination between regulatory authorities, scientific societies and patient organisations, together with biomedical literacy strategies[19].
Finally, there is a structural lack of trained professionals in this field[4]. The interdisciplinary nature of ATMPs, ranging from manufacturing and regulation to pharmacovigilance and clinical care, requires integrated profiles that are in short supply today. Encouraging joint training programs between universities, clinical centers, regulatory agencies and industry will be fundamental to consolidate a solid ecosystem.
Emerging solutions and future prospects
The response to these challenges is already underway. Process automation with semi-closed systems allows production to be scaled up with greater safety and reproducibility[6]. Decentralised manufacturing under schemes such as hospital exemption has proven effective for personalised, low-volume therapies, reducing costs and time without sacrificing efficacy or safety, as demonstrated by the ARI-000 model[15].
Digital and analytical technologies are also revolutionising quality control. AI-based tools enable real-time process monitoring, failure prediction and optimisation of critical cell culture variables[20], concepts aligned with Bioprocessing 4.0, in which platforms such as automated bioreactors can be used to simulate GMP processes[21]. At the same time, the use of in-line and at-line sensors and advanced analysis platforms, such as next-generation sequencing or mass cytometry, will gradually replace destructive testing, offering more accurate and less invasive control of ATMP production.
In the regulatory sphere, there are initiatives by the EMA and the AEMPS to facilitate early dialogue between non-commercial developers and regulatory authorities, thereby accelerating the transition to clinical trials and conditional authorisations. At the same time, programmes such as the EMA’s PRIME[22] and the FDA’s RMAT[23] prioritise the development of ATMPs for serious diseases with no therapeutic alternatives, offering fast-track assessment and approval routes. Collaborative network initiatives such as the Spanish Network for Advanced Therapies[24] (TERAV+, in Spain), the Dutch infrastructure for cancer-specific ATMP Research[25] (DARE-NL, in the Netherlands) and GoCAR-T[26] (at European level) are creating shared ecosystems that allow production to be scaled up in hospitals without relinquishing public control [27]
The creation of interoperable registries, promoted by the EBMT, will enable proactive pharmacovigilance and decisions based on real evidence. Finally, scientific societies such as the International Society for Cell Therapy (ISCT) and the EBMT are leading the way in training professionals through specialised programmes and conferences, thereby building the talent needed for the future of the field.

Figure 2. Emerging solutions and future prospects
Conclusions
ATMPs are no longer a promise for the future but have become a tangible clinical reality. However, their real impact will depend on the ability to reach those who need them most in a timely, safe and equitable manner. To achieve this, in addition to technical innovations, it is necessary to rethink and redesign the institutional frameworks that support their development, approval and clinical application.
For advanced therapies to become a structural part of 21st-century medicine, a systematic approach is needed to move from exceptional cases to standard care. This involves committing to decentralised models, adaptive regulations, collaborative clinical networks and funding aligned with value and results.